
















































服务电话
400-600-5039

精微高博公众号
北京精微高博仪器有限公司
Sales@jwgb.net
 比表面积分析仪
介孔孔径分析仪微孔孔径分析仪化学吸附仪反应评价装置蒸汽吸附仪穿透曲线分析仪真密度仪压汞仪高压吸附仪热分析仪X射线衍射仪脱气机质谱仪相关配件
比表面积分析仪
介孔孔径分析仪微孔孔径分析仪化学吸附仪反应评价装置蒸汽吸附仪穿透曲线分析仪真密度仪压汞仪高压吸附仪热分析仪X射线衍射仪脱气机质谱仪相关配件 化学吸附仪
蒸汽吸附仪
穿透曲线分析仪
真密度仪
压汞仪
脱气机
质谱仪
相关配件
化学吸附仪
蒸汽吸附仪
穿透曲线分析仪
真密度仪
压汞仪
脱气机
质谱仪
相关配件

paper
Peng Jin, Wenlong Tan, Jia Huo*, Tingting Liu, Yu Liang, Shuangyin Wang,*and Darren Bradshaw*
S1. Experimental Section
S2. Preparation and characterization of interfacial nanoassembly/ emulsion polymerization
S3. Generality of interfacial nanoassembly/ emulsion polymerization
S4. Knoevenagel condensation reaction catalyzed by ZIF-8-based catalysts
S1. Experimental Section
Materials: All chemicals were purchased from Sinopharm Chemical Reagent Co. Ltd, China and used without pretreatment unless otherwise mentioned. ZIF-8, UiO-66, Cr-MIL-101, and HKUST-1 were synthesized according to previous reports1, 2 or with minor modification. Fe3O4 nanoparticles with an average dimeter of 10 nm were obtained commercially and used without modification.
Synthesis of ZIF-8/PS: Typically, 0.70 g ZIF-8 was dispersed in 6 g deionized (DI) water using ultrasound for 30 min. 0.0761 g of 2,2'-azobisisoheptonitrile (ABVN) was dissolved in a mixture of 3.3 g divinylbenzene (DVB), 2.2 g styrene, and 0.36 g oleic acid, which was subsequently mixed with the ZIF-8 dispersion in a 20 ml sample vial. A stable Pickering emulsion was generated by agitation using a homogenizer at 14500 rpm for 2.5 min. The emulsion was then subsequently polymerized at 65 °C for 20 min. The obtained monolith was repeatedly washed with ethanol and s dried at 120 °C under vacuum. For the magnetic ZIF-8/PS, 0.07g of Fe3O4 and 0.63 g ZIF-8 were suspended into DI water using ultrasound prior to addition of the monomer solution as for ZIF-8/PS.
Synthesis of UiO-66/PS: Typically, 0.50 g UiO-66 was dispersed in 4 g of DI water by ultrasound for 30 min. 0.0761 g of ABVN was dissolved in a solution of 3.3 g DVB and 2.2 g styrene, and then the two solutions were subsequently mixed in a 20 ml sample vial. A stable Pickering emulsion was generated by agitation using the homogenizer at 14500 rpm for 2.5 min. The resultant emulsion was subsequently polymerized at 65 °C for 20 min. The obtained monolith was repeatedly washed with ethanol and subsequently dried at 120 °C under vacuum.
Synthesis of Cr-MIL-101/PS: Typically, 0.0817 g of ABVN was dissolved in the solution of 4.595 g DVB and 0.988 g 4-vinylpyridine. 0.50 g Cr-MIL-101 was dispersed in 4 g DI water by ultrasound for 6 h after which time 0.2 g NaCl was added into it, and then the two solutions were mixed with strong stirring (1000 rpm) in a 20 ml sample vial. A stable Pickering emulsion was generated by agitation using the homogenizer at 14500 rpm for 2.5 min. The resultant emulsion was subsequently polymerized at 65 °C for 20 min. The obtained monolith was repeatedly washed with ethanol and subsequently dried at 120 °C under vacuum.
Synthesis of HKUST-1/PAM: 0.30 g HKUST-1 was dispersed in 6 mL xylene by ultrasound for 6 h. Acrylamide (AM, 1.533 g), N,N′-methylenebisacrylamide (MBAM, 0.311 g), and N-isopropylacrylamide (NIPAM, 0.40 g) were dissolved in 4 g of distilled water to obtain the monomer aqueous solution. The HKUST-1 dispersion was added slowly into the monomer aqueous solution followed by addition of 0.2 g of 4,4'-azobis(4-cyanovaleric acid) (ACVA) during stirring (1000 rpm). Stirring was continued for a further 1 min after addition. A stable Pickering emulsion was generated with agitation using the homogenizer at 14500 rpm for 2.5 min. The resultant emulsion was transferred to a preheated oven at 60 °C for 20 min. After removal from the oven, the obtained monolith was continually washed with acetone using a Soxhlet extractor for 24 h and then dried under vacuum at 120 °C overnight.
Characterization: A stable Pickering emulsion was generated using the homogenizer T10 B S25 (IKA) at 14500 rpm for 2.5min.The phase identification of the samples was performed on a Powder X-ray diffractometer (XRD, D8 Advance) using Cu Kα radiation, where the scanning range of the diffraction angle (2-Theta) is 5-50 °. The scanning rate was 4 ° min-1 and step width of 0.02 ° with 40 mA operation current and 40 kV voltage. Thermogravimetric analysis (TGA) was performed using a HTG-1 (HENVEN) instrument and the sample was heated from room temperature to 800 oC at a rate of 10 oC min-1 under an air atmosphere. Scanning electron microscopy (SEM) measurements were made on a Hitachi S-4800 thermal field emission scanning electron microscope at an accelerating voltage of 5, 10, or 15 kV. Samples for SEM measurements were attached to the stub using carbon paste and then sputter-coated with a thin layer of conductive gold to improve electrical conductivity. The nitrogen adsorption-desorption isotherms were measured at 77 K with a JW-BK200C (Beijing JWGB Sci.&Tech. Co., Ltd.) gas adsorption analyser after the sample was first degassed at 120 °C overnight. Specific surface areas were determined by the BET method, mesopore size distribution was obtained based on BJH analysis of the desorption branches of the isotherms, and the total pore volumes were determined using the desorption branch of N2 isotherm at p/p0 = 0.99 (single point). Gas chromatographic (GC) analysis was performed using a Lunan Ruihong SP-7820 instrument equipped with a flame ionization detector (FID) and an SE-54 column (length = 30 m, inner diameter = 0.32 mm, and film thickness = 0.50 μm). The temperature for GC analysis ramped from 60 to 140 oC at 20 oC min-1 and was held at 140 oC for 1 min; then was heated from 140 to 142 at 1 oC/min; and then ramped to 280 oC at 20 oC min-1 and was held them at 280 oC for 2 min. Inlet and detector temperatures were set at 280 oC. n-Dodecane was used as an internal standard to calculate reaction conversion.
Catalytic assessment: The Knoevenagel reaction between malononitrile and benzaldehyde using the ZIF-8/PS composite as the catalyst was performed in a round bottom flask with magnetic stirring. Typically, a mixture of 0.158 g ZIF-8/PS with 0.2 mL (1.96 mmol) benzaldehyde and 0.2 mL n-dodecane as an internal standard was placed into a 25 mL flask containing 4 mL of toluene. The solution of 0.25 g (3.78 mmol) malononitrile in 1 mL toluene was subsequently added into the flask, and the resultant mixture was stirred for 12 h at room temperature. The sample was taken at specific time intervals and analyzed by GC. After the reaction, the catalyst was removed from the reaction mixture, washed with ethanol for three times, and dried under vacuum at 120 °C for 8 h prior to being reused.
S2. Preparation and characterization of ZIF-8/PS via Interfacial Nanoassembly/ Emulsion Polymerization
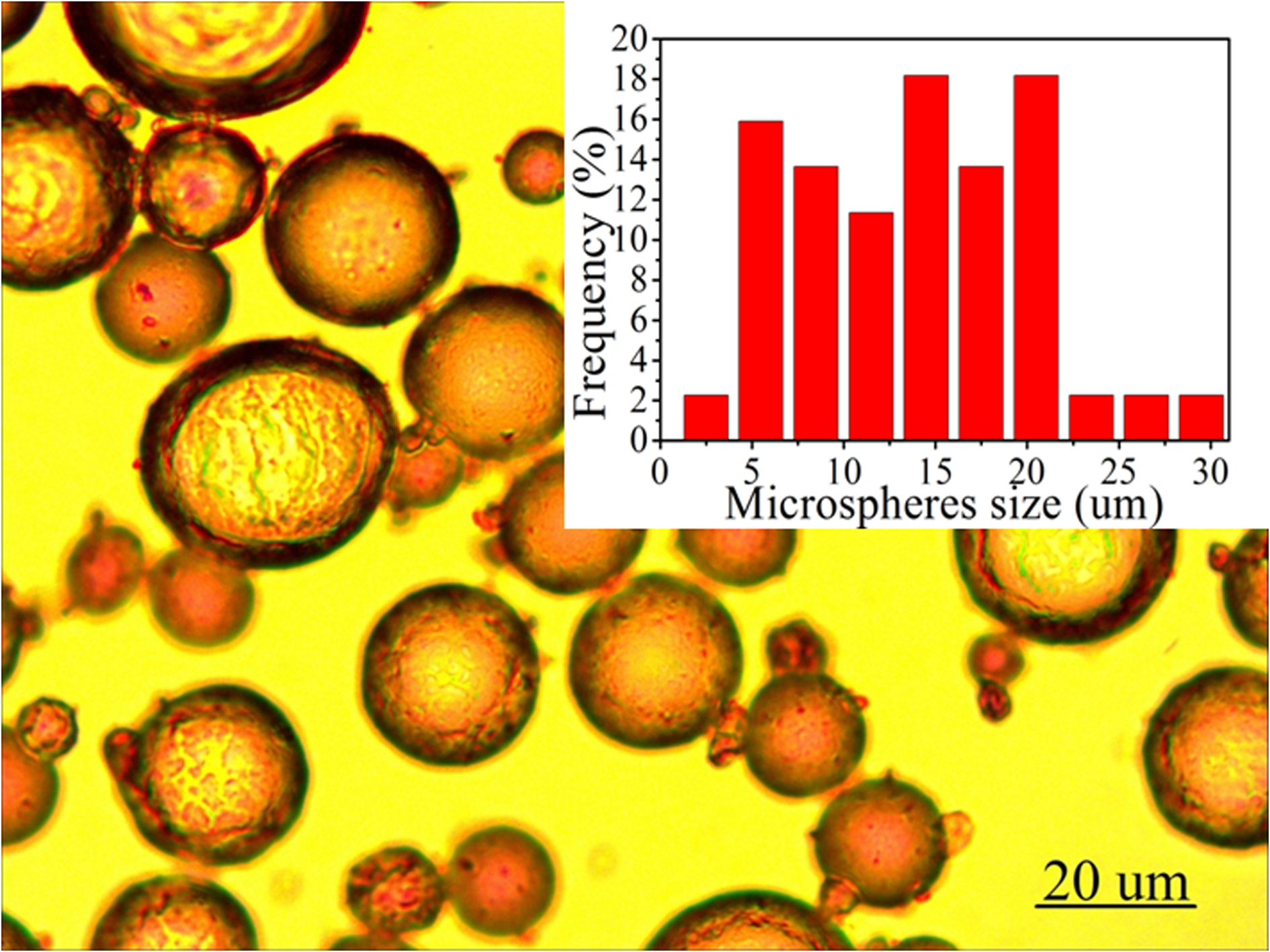
Figure S1. Optical microscope image of ZIF-8-stablized emulsions employed for the preparation of ZIF-8/PS composites through polymerization of the continuous phase. The inset shows the size distribution of the emulsion droplets which ultimately form the macropores within the composites.
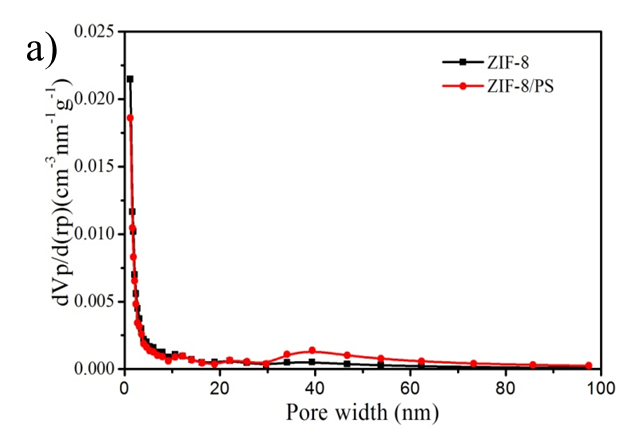
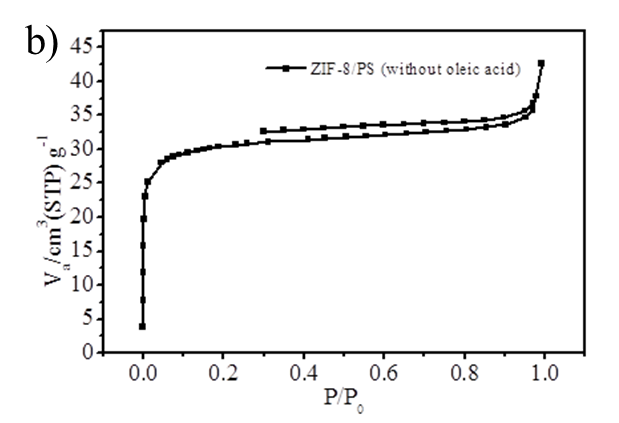
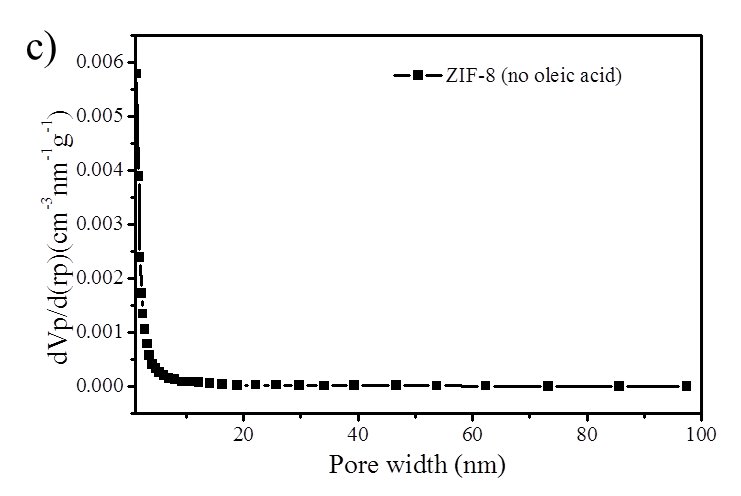
Figure S2. (a) BJH mesopore size distribution of ZIF-8 nanoparticles and hierarchically porous ZIF-8/PS composites prepared with oleic acid; (b) N2 isotherm (Specific surface area is 118 m2/g) and (c) mesopore size distribution of ZIF-8/PS prepared without oleic acid demonstrating a reduction in surface area and an absence of mesopores.


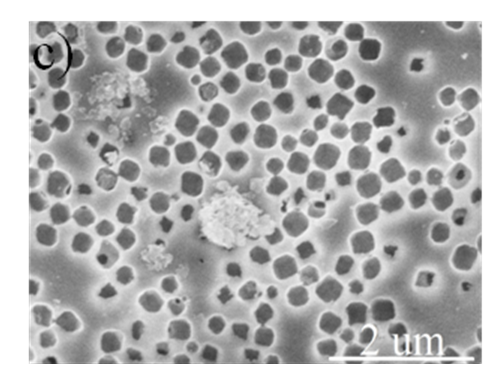
Figure S3. Low and high magnification SEM images of the cross-section of hierarchically porous ZIF-8/PS after removal of the ZIF-8 particles by acetic acid dissolution, further confirming their localisation within the macropores.
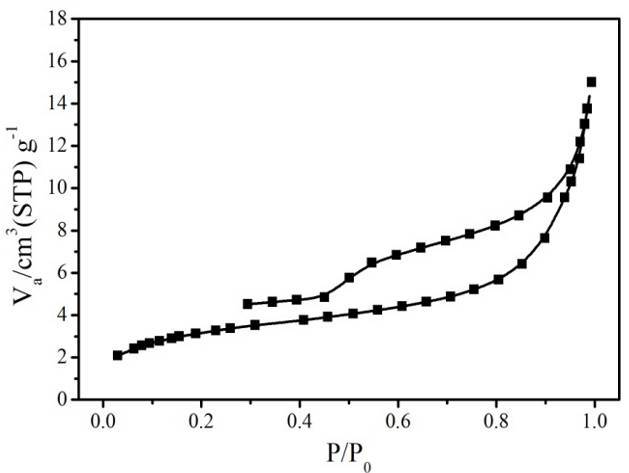
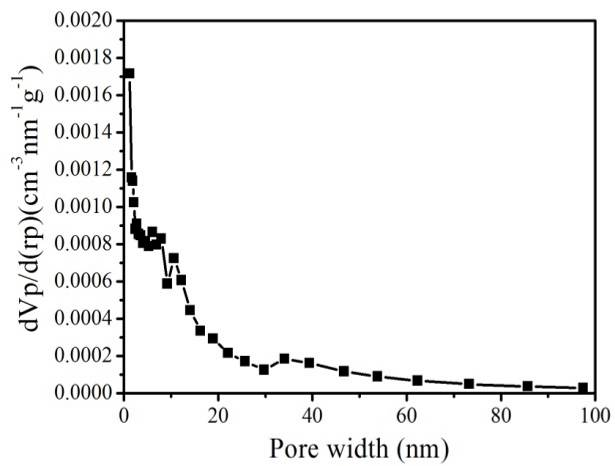
Figure S4. N2 sorption isotherm (top) and pore-size distribution (bottom) of hierarchically porous ZIF-8/PS after removing the ZIF-8 by acetic acid dissolution.
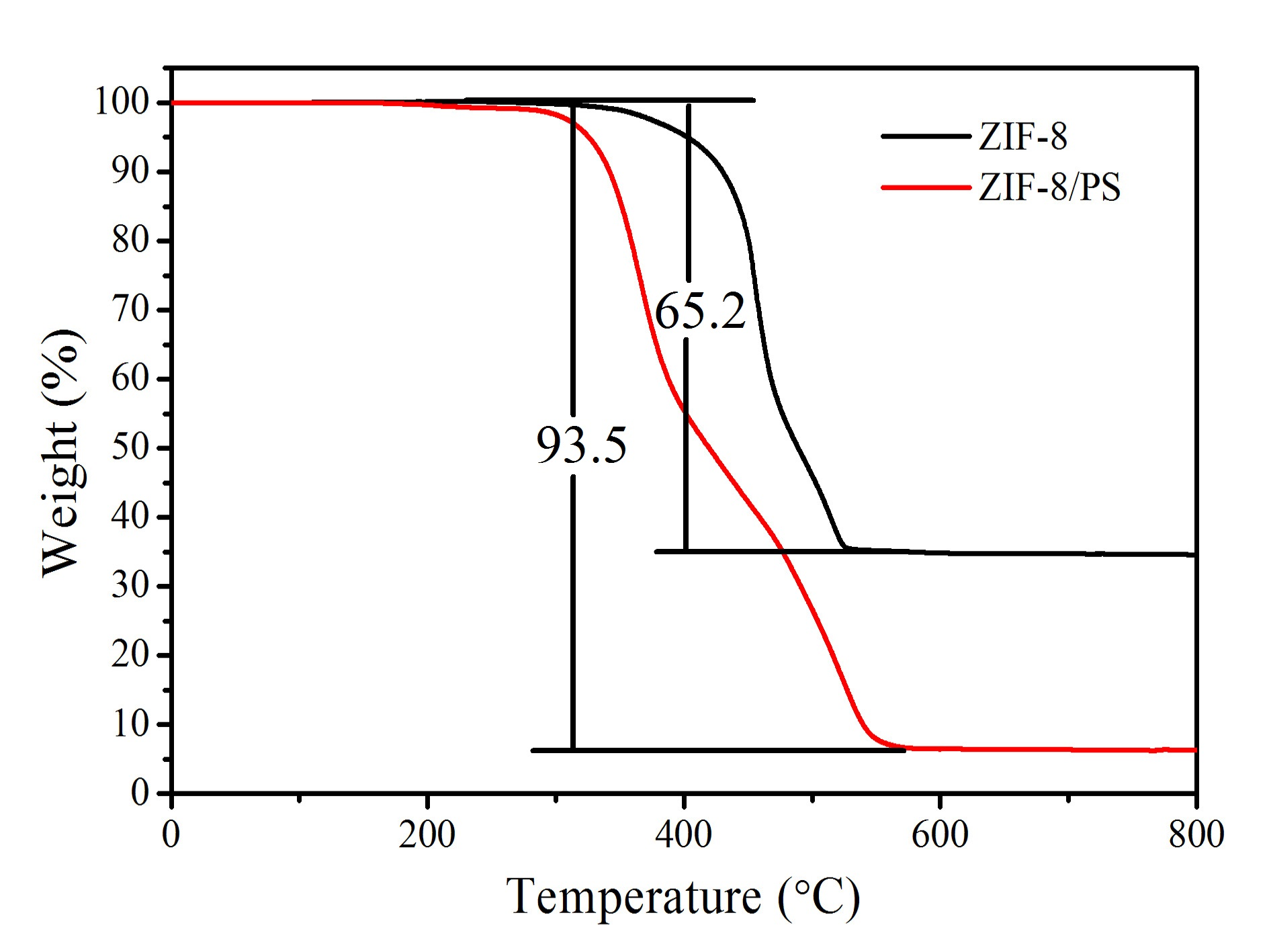
Figure S5.TGA data of ZIF-8 nanoparticles and hierarchically porous ZIF-8/PS composites.
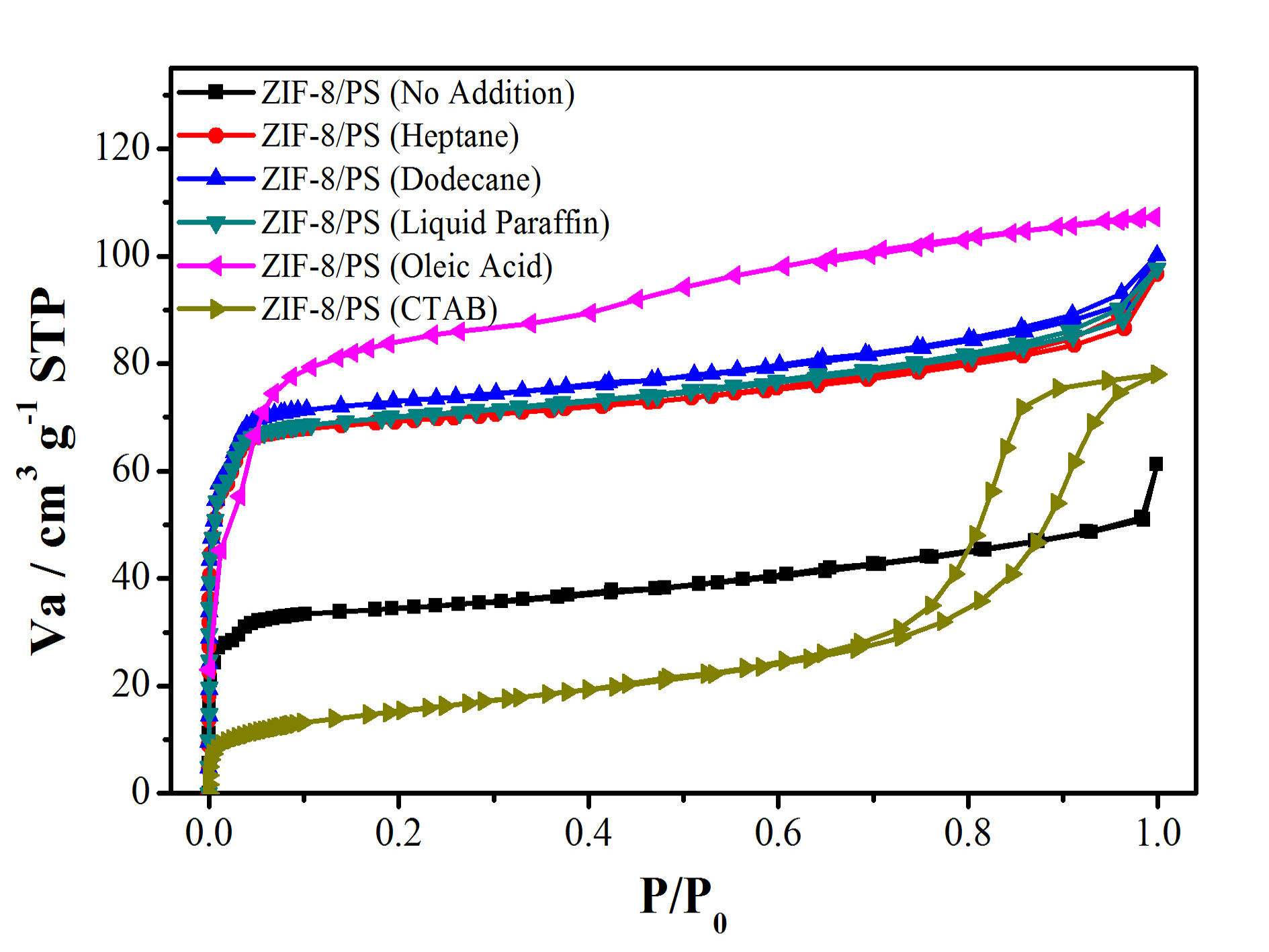
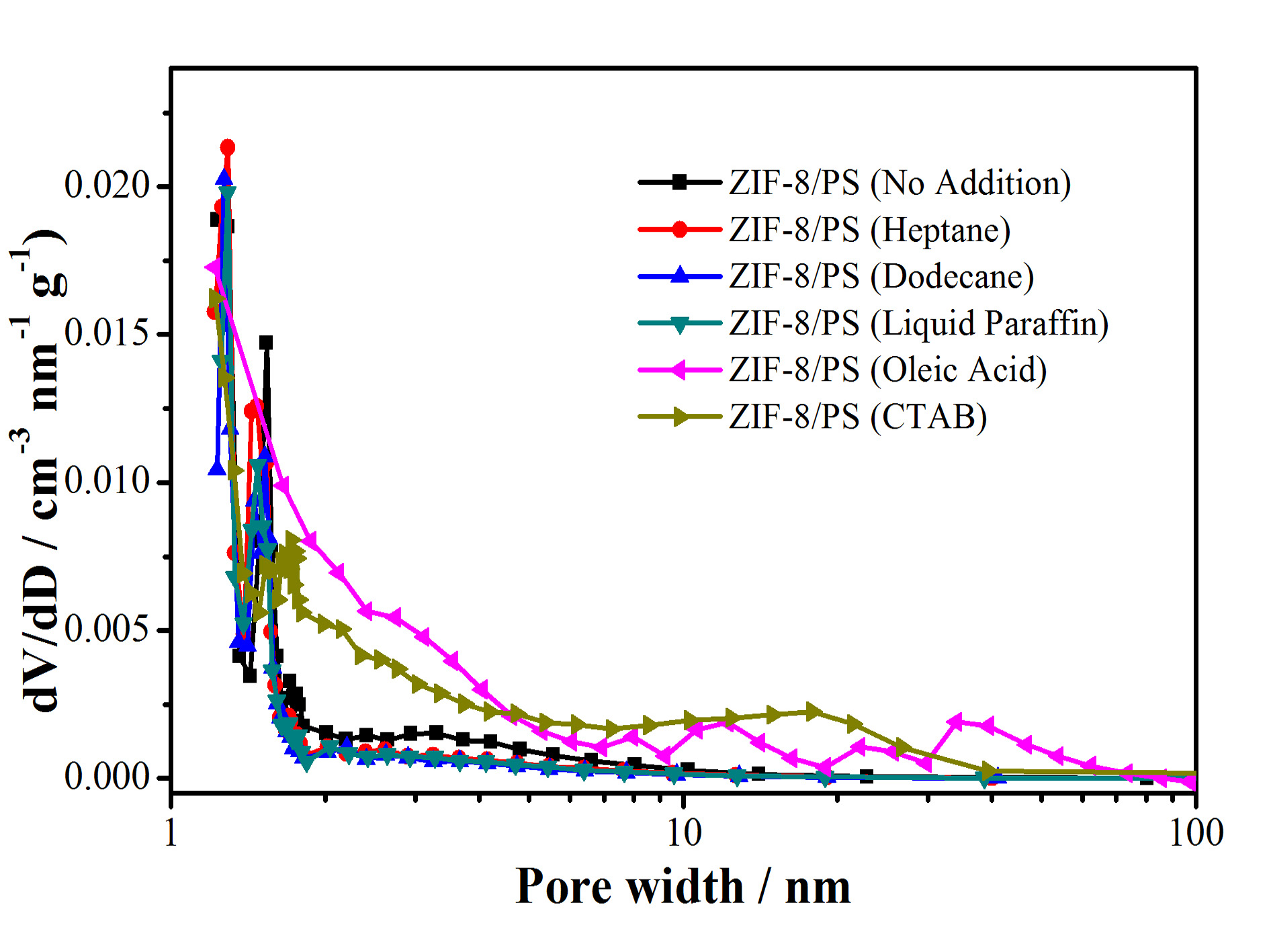
Figure S6. N2 sorption isotherm (top) and pore-size distribution (bottom) of hierarchically porous ZIF-8/PS prepared without or with different swelling agents .
Table S1. Specific surface area (BET method) of hierarchically porous ZIF-8/PS prepared without or with different swelling agents.
swelling agents | no addition | heptane | dodecane | liquid paraffin | oleic acid | CTAB |
BET surface area | 133 | 275 | 276 | 279 | 299 | 178 |
1. J. Huo, M. Marcello, A. Garai and D. Bradshaw, Adv. Mater., 2013, 25, 2717-2722.
2. L. H. Wee, M. R. Lohe, N. Janssens, S. Kaskel and J. A. Martens, J. Mater. Chem., 2012, 22, 13742-13746.
服务电话
精微高博公众号
Sales@jwgb.net
 京公网安备 11011202004400号
京公网安备 11011202004400号